Attention: The Herranz Lab is now hiring! Click here for more information.
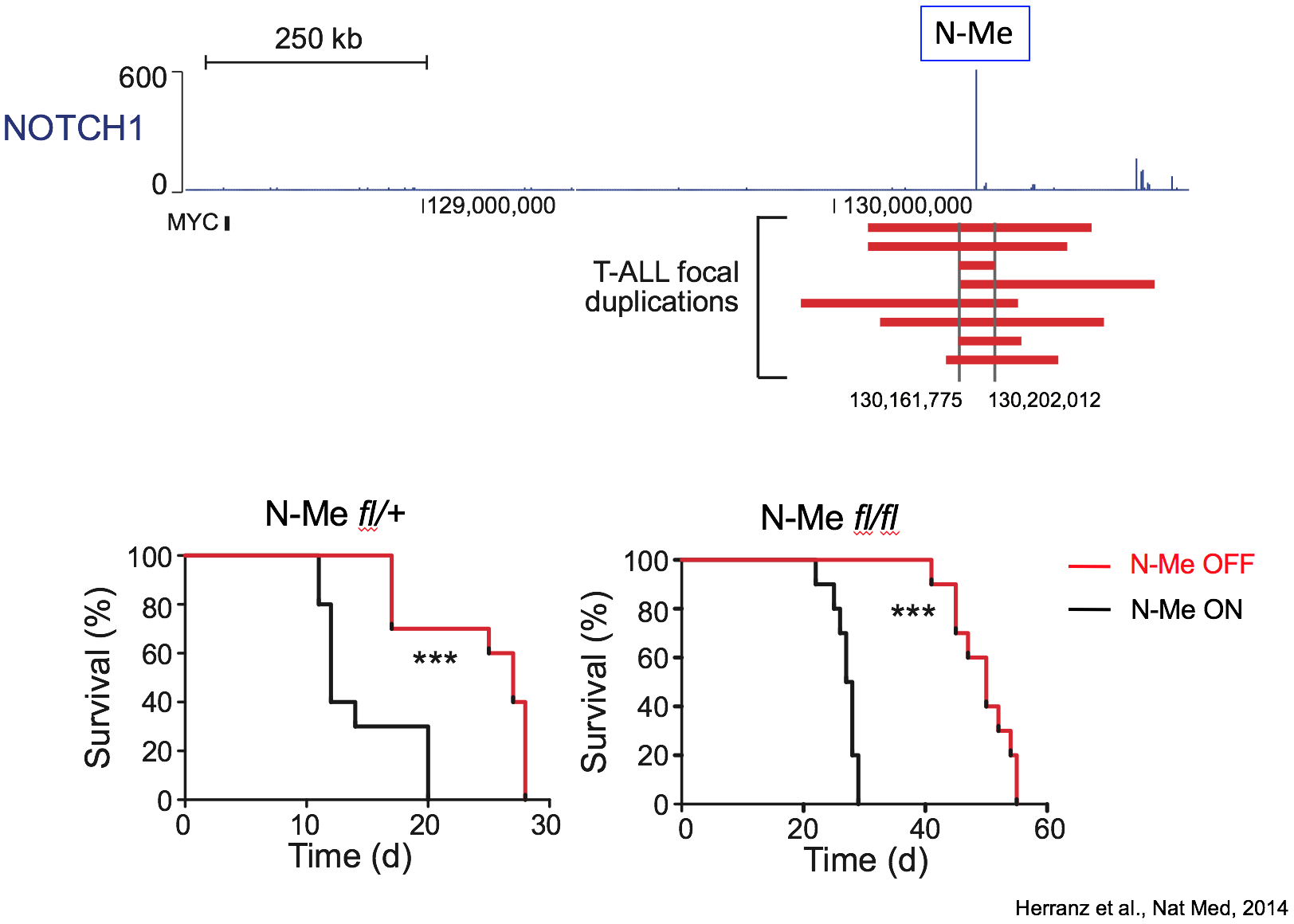
N-Me: an oncogenic enhancer in NOTCH-induced T-ALL
The Herranz lab was established in July 2017 at the Rutgers Cancer Institute and our long-term research goals are: (i) to discover and define how oncogenic and tumor suppressor enhancers impact tumorigenesis in hematological malignancies; (ii) to dissect the interplay between cancer cell-specific metabolic rewiring and epigenetics in T-cell Acute Lymphoblastic Leukemia (T-ALL).
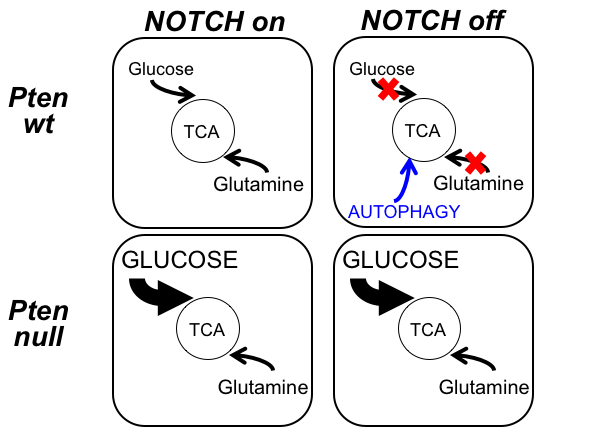
Metabolic reprogramming of T-ALL cells confers resistance to NOTCH inhibition
The first area of interest in the lab stems from our initial discovery of a Notch1-bound Myc enhancer (N-Me), as the long-sought missing link in the regulation of MYC by NOTCH1 in T-cells. Using an integrated analysis of human and mouse NOTCH1 ChIP-seq data, chromosome conformation capture analyses, reporter assays and novel mouse models, we demonstrated that N-Me loops with the MYC promoter in T-cells, inducing Myc expression in an orientation-independent but NOTCH1-dependent-manner. Moreover, experiments using N-Me knockout and conditional knockout mouse models showed that N-Me is critically required for the normal development of T-cells, as well as for the generation and maintenance of NOTCH1-induced T-ALL (Herranz et al, Nature Medicine 2014).
Our central hypothesis is that enhancer malfunction is a recurrent and significant hallmark of cancer in different tumor types. Moreover, we hypothesize that in addition to duplications of oncogenic enhancers, there will also occur deletions of tumor suppressor enhancers that control the expression of haploinsufficient tumor suppressor genes. Thus, our main goal is to discover and dissect novel oncogenic and tumor suppressor enhancers.
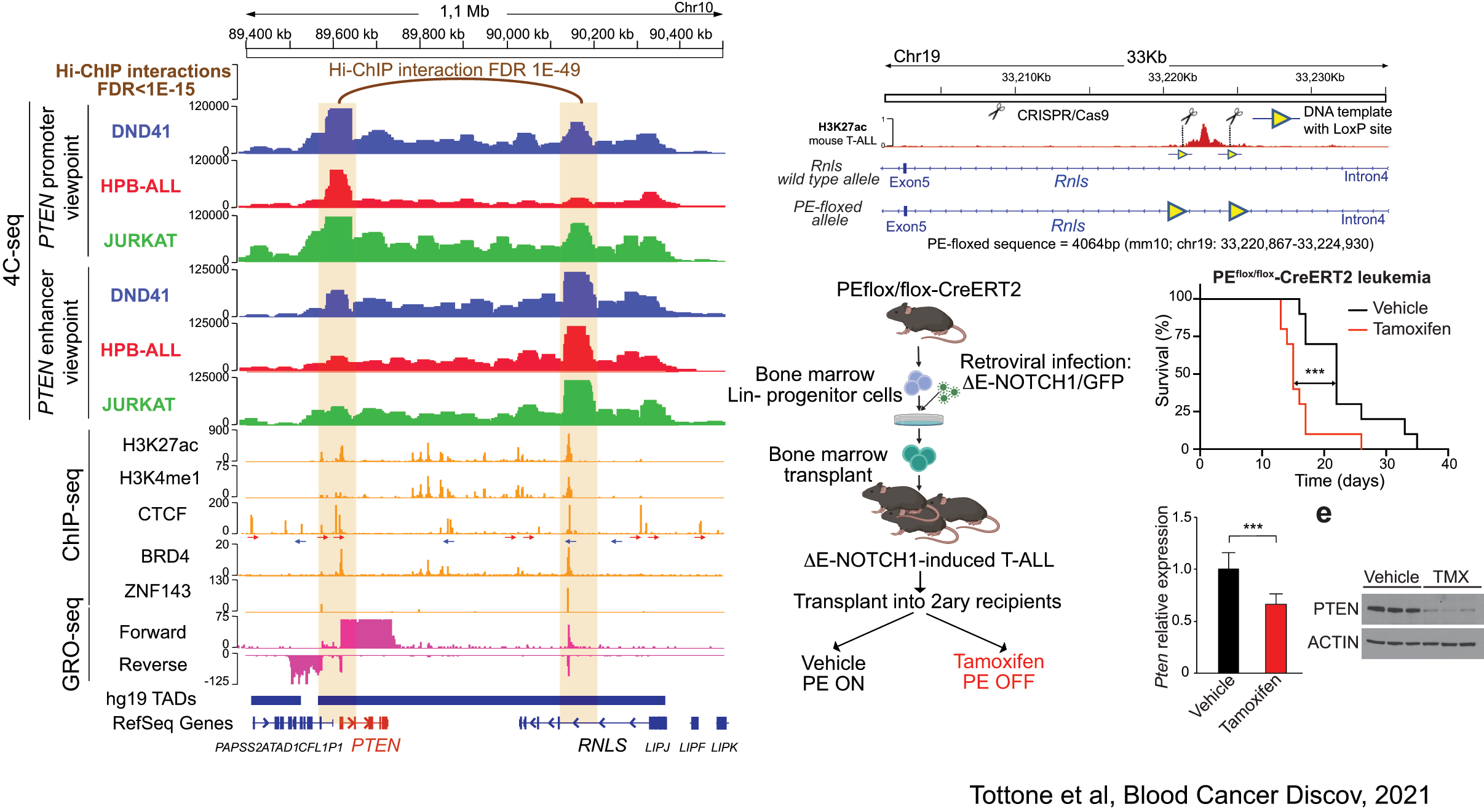
PE: a tumor suppressor enhancer of PTEN in T-ALL
Related to this, our lab has recently discovered the first enhancer of a major tumor suppressor gene. Indeed, using a combination of 4C-seq, epigenetic profiling, scRNA-sequencing and novel mouse models, we have identified a tumor supressor enhancer of PTEN that plays a relevant role in T-ALL, and which is recurrently deleted in T-ALL patients (Tottone et al, Blood Cancer Discov 2021).
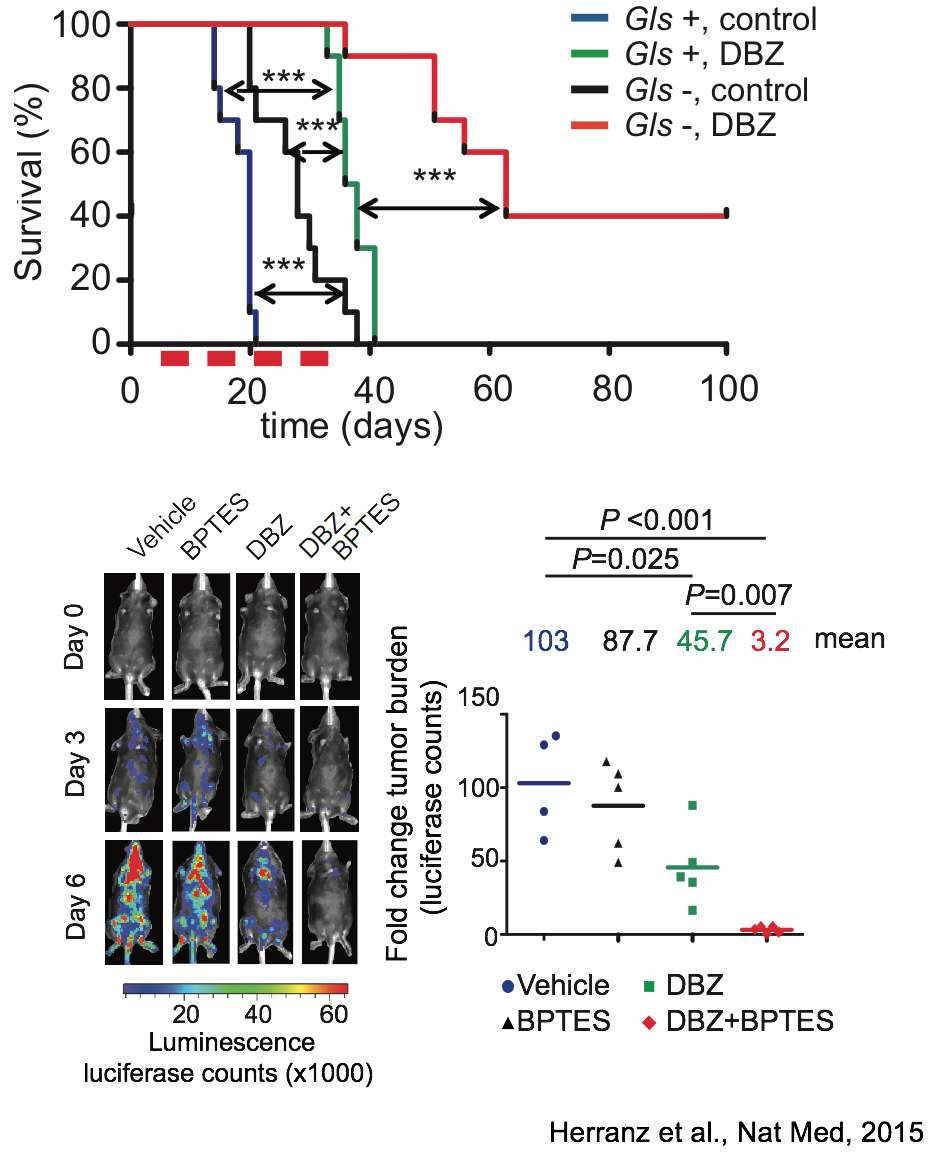
Synergistic antileukemic effects of NOTCH and glutaminolysis inhibition in T-ALL
The second area of interest in the lab sprouts from our long-standing interest in the interplay between metabolism and epigenetics as a pathogenic mechanism of cancer. In particular, using a combination of gene expression profiling, metabolomic analyses, 13C isotope labeling experiments and state-of-the-art experimental therapeutics in vivo, we uncovered a critical role for the metabolic effects of NOTCH1 inhibition in the antileukemic activity of anti-NOTCH1 therapies. Specifically, these studies revealed that inhibition of NOTCH1 signaling elicits a global anabolic shutdown, forcing leukemic cells to use autophagy as a salvage pathway to support cell growth and demonstrated that these effects are rescued by isogenic loss of Pten. Moreover, these studies revealed a prominent role of glutaminolysis in feeding the tricaboxylic acid downstream of NOTCH1 in Pten-positive T-ALL. Consistent with these findings, autophagy inhibition or glutaminolysis inhibition, either by genetic or pharmacological means, show highly synergistic antileukemic effects with anti-NOTCH1 therapies in vivo (Herranz et al, Nature Medicine 2015).
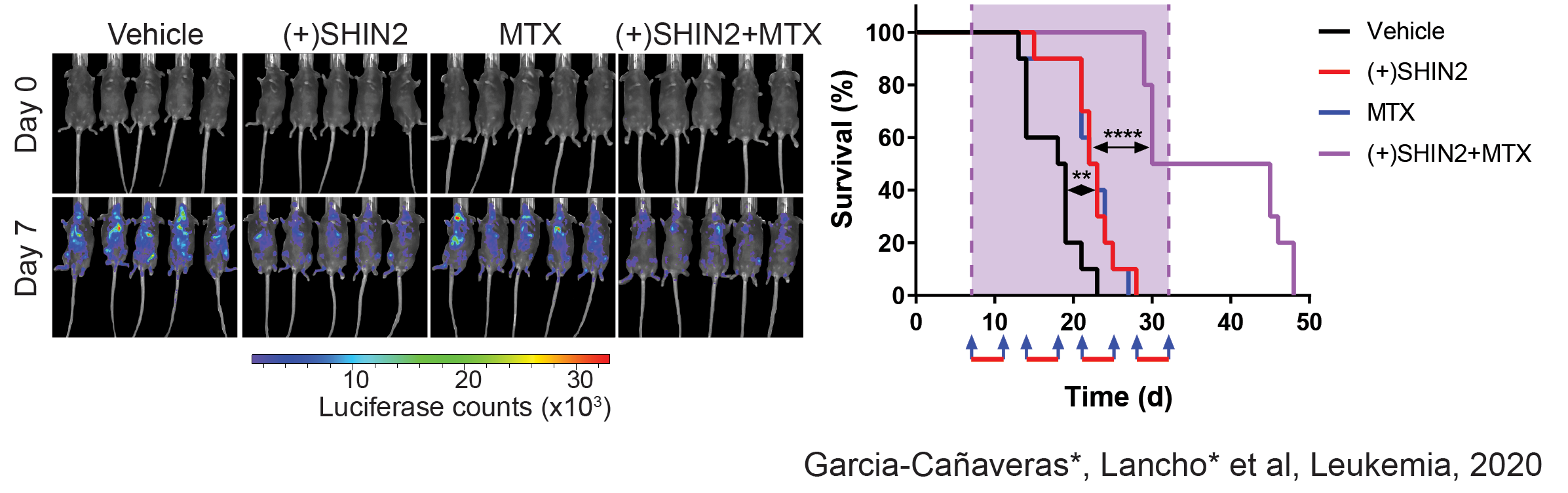
Serine Hydroximethyl transferase inhibition with SHIN2 shows antileukemic and highly synergistic effects with methotrexate in vivo
We have recently made new discoveries in this are that open new options for therapeutic treatment. Indeed, in a joint collaboration with the Rabinowitz Lab at Princeton, we have identified SHMT as a relevant therapeutic target in T-ALL, and showed that SHMT inhibition in vivo leads to very significant antileukemic effects and synergizes with Methotrexate treatment (García-Cañaveras*, Lancho* et al, Leukemia 2020).In addition, we have identified the highly antileukemic effects of mitochondrial uncoupling in vivo. Specifically, MB1-47 (a novel niclosamide-based uncoupler with much improved exposure) leads to broad and AMPK-mediated antileukemic effects in different mouse primary leukemias and human T-ALL PDXs (da Silva-Diz et al, Blood 2021)
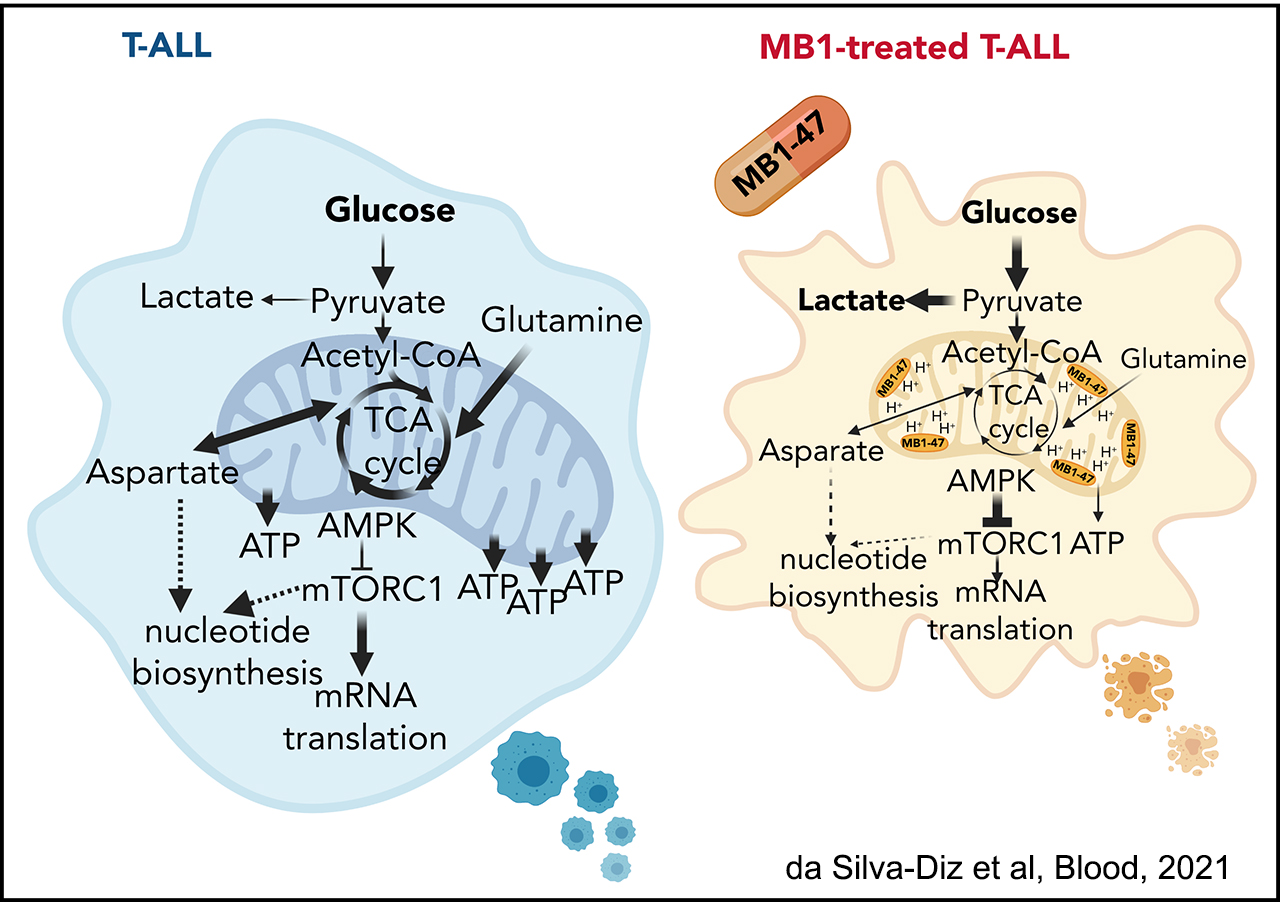
The novel mitochondrial uncoupler MB1-47 shows broad antileukemic effects in vivo
Moreover, we recently identified a novel axis in T-ALL in which NOTCH1 transcriptionally activates SIRT1 via an enhancer region, and SIRT1 in turn deacetylates and activates the histone acetyltransferase KAT7. As such, both SIRT1 and KAT7 are novel interesting targets for T-ALL treatment (Lancho et al, Blood Cancer Discov 2023)

A novel NOTCH1-SIRT1-KAT7 axis in T-ALL
Moving forward, we hypothesize that key regulators that affect both the epigenetic and metabolic status of cancer cells will act as master regulators of oncogenic transformation. As such, we will test the effects of different candidates on the metabolic and epigenetic landscape T-ALL in vivo. These experiments will open new therapeutic opportunities for the treatment of this disease.
Click here for a comprehensive list of publications.